About the Martini Force Field Initiative
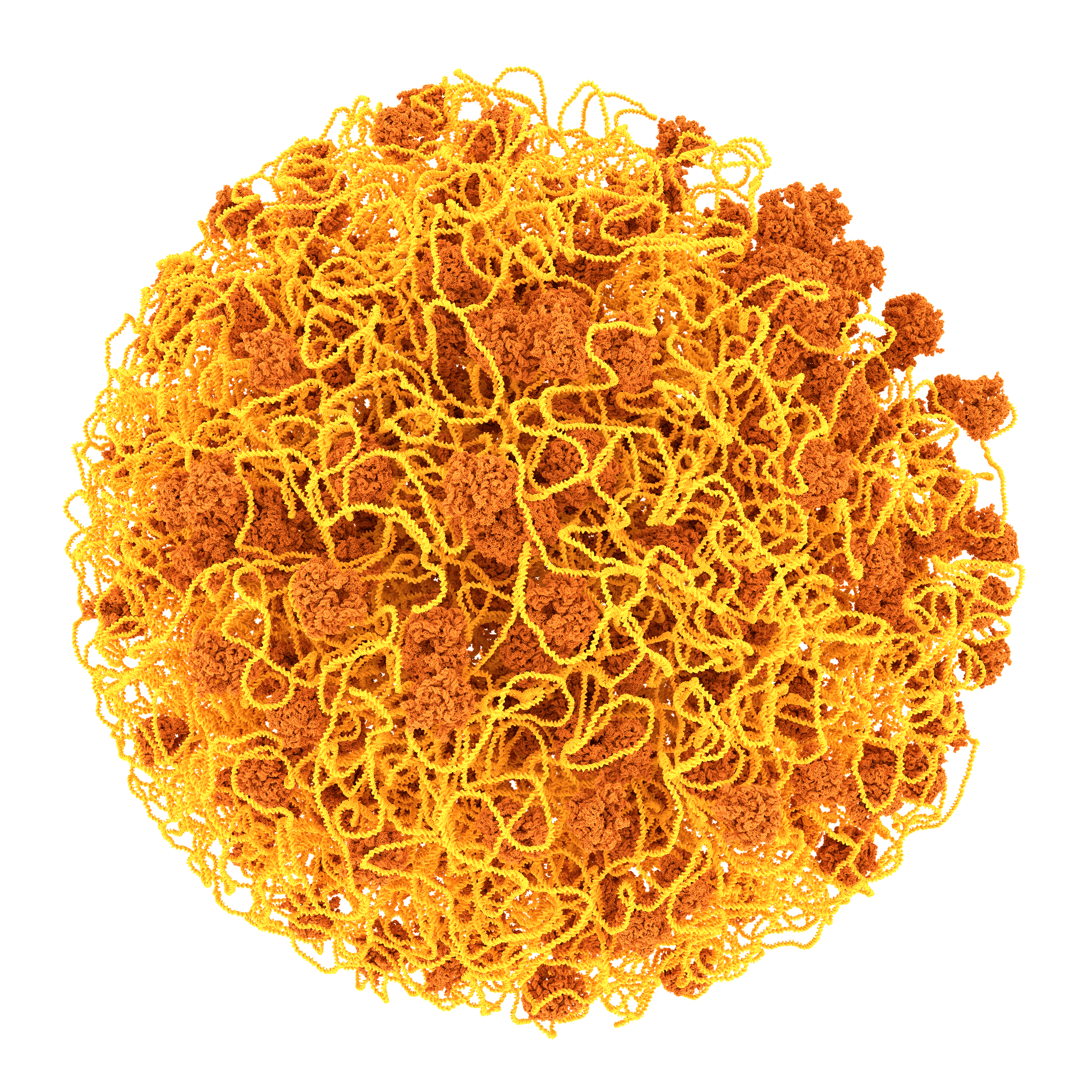
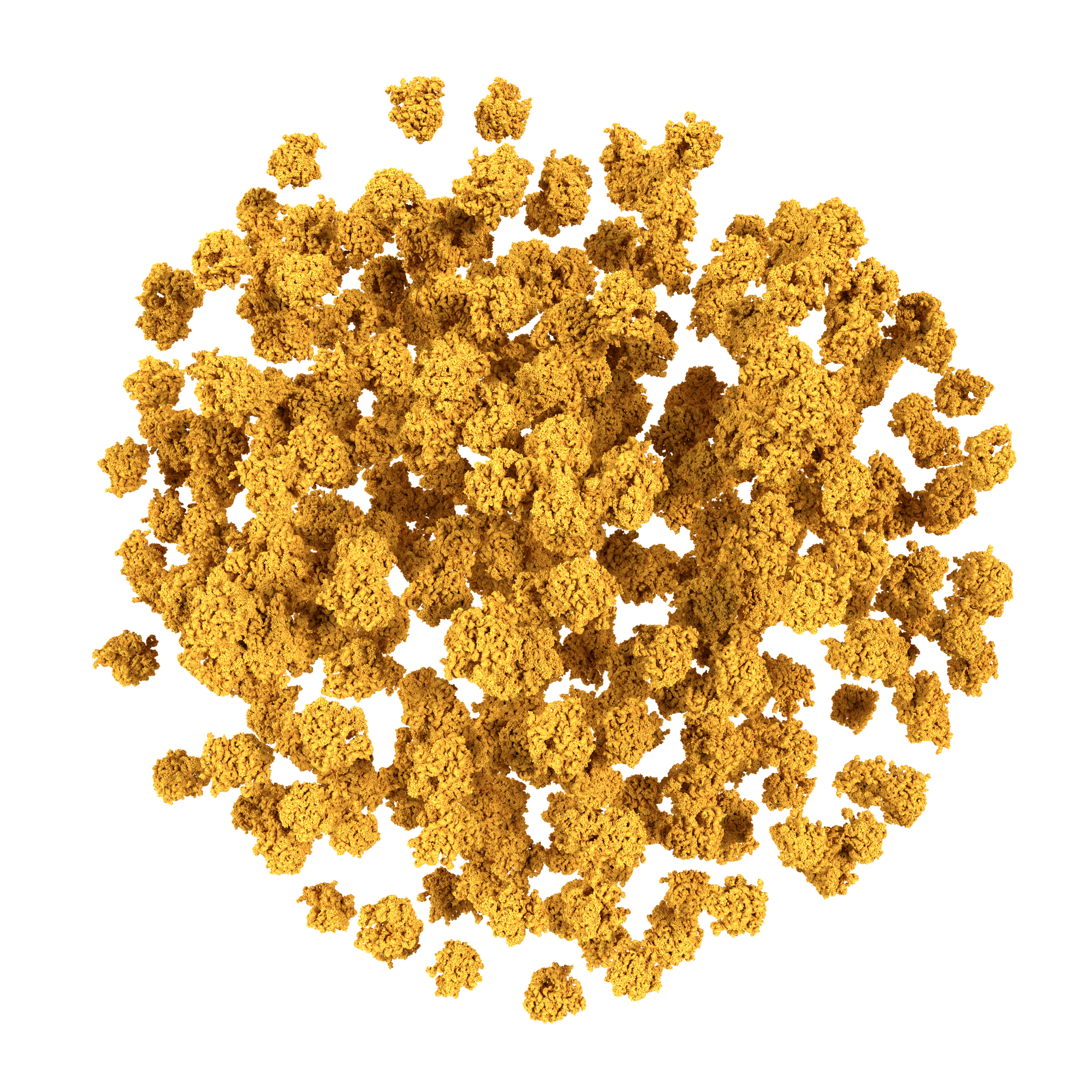
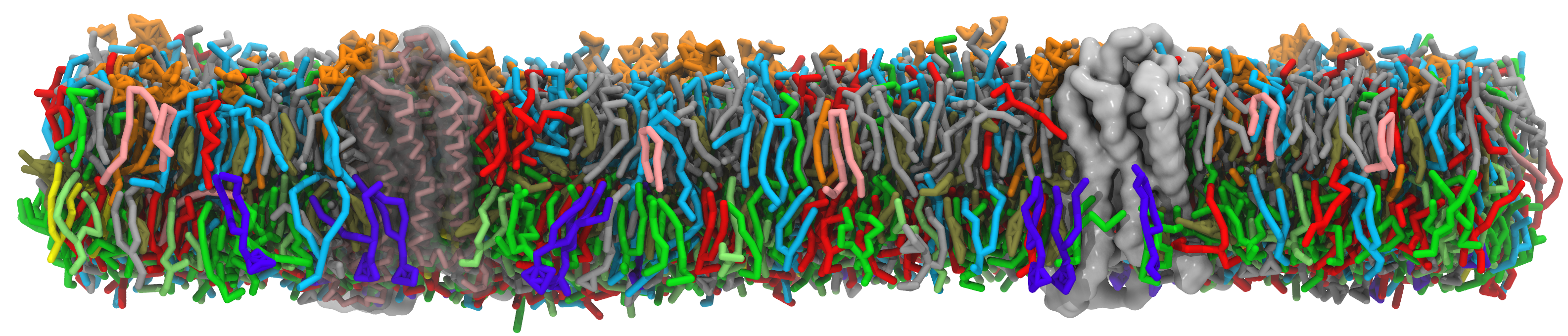
Martini is a coarse-grained (CG) force field suited for molecular dynamics simulations of (bio)molecular systems. The force field has been parametrized in a systematic way, combining top-down and bottum-up strategies: i- non-bonded interactions are based on the reproduction of experimental partitioning free energies between polar and apolar phases of a large number of chemical compounds, whereas ii- bonded interactions are derived from reference all-atom simulations.
The model uses a four-to-one mapping scheme, i.e. on average four heavy atoms and associated hydrogens are represented by a single interaction center. In order to keep the model simple, only four main types of interaction sites are defined: polar, non-polar, apolar, and charged. Each particle type has a number of subtypes, which allow for an accurate representation of the chemical nature of the underlying atomistic representation.
Currently topologies are available for many lipids and surfactant molecules, including cholesterol, and for all amino acids and nucleotides as well as for a variety of sugars, polymers, and nanoparticles. Tools are furthermore available to build topologies for arbitrary peptides, proteins, polynucleotides, to add elastic networks, and to move between coarse-grained and atomistic representations.